![]()
![]()
![]()

第Ⅲ相臨床試験 ③(腎機能障害を伴う2型糖尿病患者における長期投与試験)
1試験概要
デザイン |
多施設共同無作為化プラセボ対照二重盲検比較試験(並行群間比較法) |
|---|---|
| 目的 | 腎機能障害を伴う2型糖尿病患者を対象に、ルセフィを52週間長期投与した際の安全性および有効性を検討する。 |
| 対象 | 腎機能障害(中等度)を伴う2型糖尿病患者145例
|
| 方法 |
ルセフィ2.5mg群、プラセボ群に無作為に割り付け、それぞれの試験薬を二重盲検下で1日1回、朝食前に24週間経口投与した(二重盲検治療期Ⅰ)。二重盲検治療期Ⅰ終了後に非盲検治療期Ⅱに移行した患者を対象に、非盲検下でルセフィ2.5mgを1日1回、28週間経口投与した。ただし、増量基準※を満たした場合は、投与24週以降にルセフィ5mg1日1回への増量を可とした。 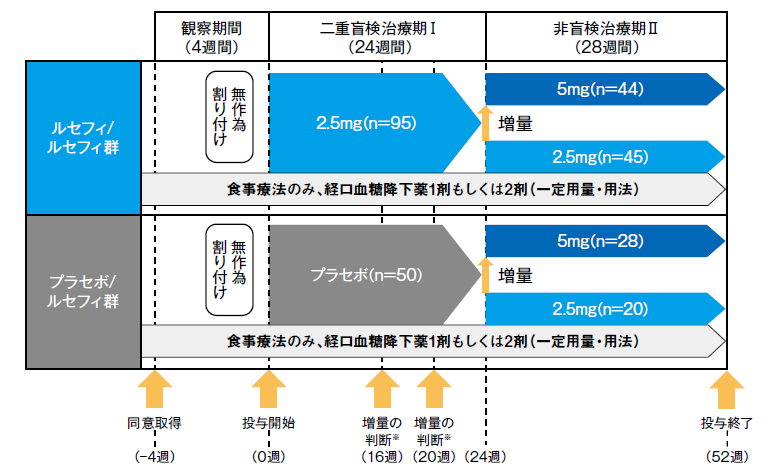
|
| 評価方法 |
治療開始時、2週、4週、以後4週ごとに52週までHbA1c、空腹時血糖値(朝食前)、体重を測定した。 |
| 評価項目 |
安全性評価項目:有害事象(臨床検査値、バイタルサイン、12誘導心電図を含む)の内容および発現頻度 有効性評価項目:HbA1c、空腹時血糖値、グリコアルブミン、体重、空腹時インスリン、血中 CPR、インタクトプロインスリン、HOMA-R、HOMA-β (HbA1c はNGSP値で表記) |
| 解析計画 | 安全性の解析対象は安全性解析対象集団とし、有効性の解析対象はFASとした。 [有効性評価項目] |
- 社内資料(臨床試験HbA1c[NGSP値]再解析結果)
- 承認時評価資料(腎機能障害を伴う2型糖尿病患者を対象とした長期投与試験)


第Ⅲ相臨床試験 ③(腎機能障害を伴う2型糖尿病患者における長期投与試験)
2HbA1cの改善効果
ルセフィ2.5mgの腎機能障害(中等度)を伴う2型糖尿病患者への投与において、二重盲検治療期Ⅰ終了時※1におけるベースラインからのHbA1c変化量は-0.11%であり、プラセボ群の0.09%に比べて、有意な低下が認められた。また、投与52週※2においても、ベースラインからのHbA1c変化量は-0.30%と、ベースラインに比べて有意な低下が認められた。
■二重盲検治療期Ⅰ終了時および52週におけるベースラインからのHbA1c変化量
二重盲検治療期Ⅰ終了時※1
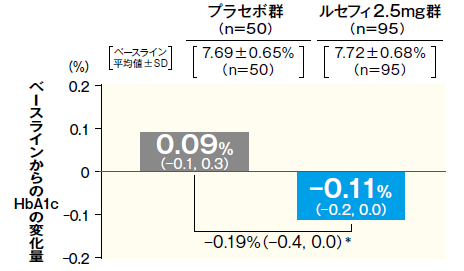
- FAS 最小二乗平均値(95% CI)
- *:p < 0.05(vs. プラセボ群)、治療期開始時(ベースライン)の値を共変量とした共分散分析
52週におけるベースラインからのHbA1c変化量
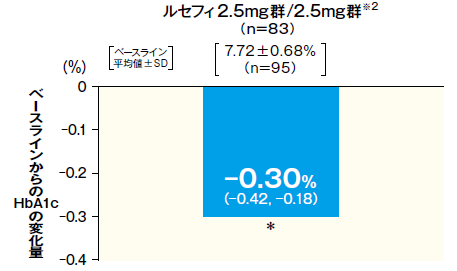
- FAS 平均値(95%CI)
- *:p < 0. 001(vs. ベースライン)、1標本t検定
- ※1 中止、脱落例などは、最終観察時点の測定値を解析。
- ※2 投与24週まで二重盲検下でルセフィ2.5mgを投与し、引き続き、非盲検下でルセフィ2.5mgを投与した症例。投与24週以降にルセフィ5mgへ増量した症例を含む。
- 8. 重要な基本的注意(抜粋)
- 8.3 本剤投与により、血清クレアチニンの上昇又はeGFR の低下がみられることがあるので、腎機能を定期的に検査するとともに、腎機能障害患者における治療にあたっては経過を十分に観察すること。[5.3 、5.4 、9.2 参照]
- 9.2 腎機能障害患者
- 9.2.1 重度の腎機能障害(eGFRが15mL/min/1.73m2以上29mL/min/1.73m2以下)のある患者又は透析中の末期腎不全患者
投与しないこと。本剤の効果が期待できないため。[5.3 、8.3 、16.6.1 参照] - 9.2.2 中等度の腎機能障害(eGFRが30mL/min/1.73m2以上59mL/min/1.73m2以下)のある患者
投与の必要性を慎重に判断すること。本剤の効果が十分に得られない可能性がある。[5.4 、8.3 、16.6.1 、17.1.3 参照]
第Ⅲ相臨床試験 ③(腎機能障害を伴う2型糖尿病患者における長期投与試験)
3空腹時血糖値の改善効果
ルセフィ2.5mgの腎機能障害(中等度)を伴う2型糖尿病患者への投与において、二重盲検治療期Ⅰ終了時※1におけるベースラインからの空腹時血糖値変化量は-6.7mg/dLであり、プラセボ群の0.5mg/dLに比べて、有意な低下が認められた。また、投与52週※2においても、ベースラインからの空腹時血糖値変化量は-14.1mg/dLと、ベースラインに比べて有意な低下が認められた。
■二重盲検治療期Ⅰ終了時および52週におけるベースラインからの空腹時血糖値変化量
二重盲検治療期Ⅰ終了時※1
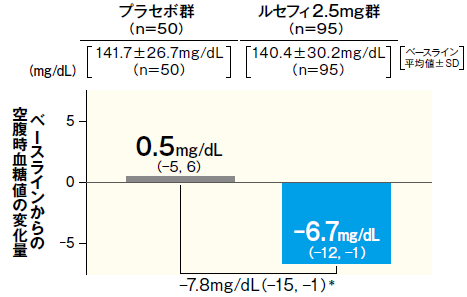
- FAS 最小二乗平均値(95% CI)
- *:p < 0.05(vs. プラセボ群)、治療期開始時(ベースライン)の値を共変量とした共分散分析
投与52週
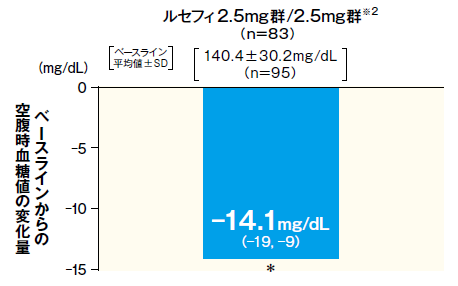
- FAS 平均値(95%CI)
- *:p < 0. 001(vs. ベースライン)、1標本t検定
- ※1 中止、脱落例などは、最終観察時点の測定値を解析。
- ※2 投与24週まで二重盲検下でルセフィ2.5mgを投与し、引き続き、非盲検下でルセフィ2.5mgを投与した症例。投与24週以降にルセフィ5mgへ増量した症例を含む。
- 8. 重要な基本的注意(抜粋)
- 8.3 本剤投与により、血清クレアチニンの上昇又はeGFR の低下がみられることがあるので、腎機能を定期的に検査するとともに、腎機能障害患者における治療にあたっては経過を十分に観察すること。[5.3 、5.4 、9.2 参照]
- 9.2 腎機能障害患者
- 9.2.1 重度の腎機能障害(eGFRが15mL/min/1.73m2以上29mL/min/1.73m2以下)のある患者又は透析中の末期腎不全患者
投与しないこと。本剤の効果が期待できないため。[5.3 、8.3 、16.6.1 参照] - 9.2.2 中等度の腎機能障害(eGFRが30mL/min/1.73m2以上59mL/min/1.73m2以下)のある患者
投与の必要性を慎重に判断すること。本剤の効果が十分に得られない可能性がある。[5.4 、8.3 、16.6.1 、17.1.3 参照]
第Ⅲ相臨床試験 ③(腎機能障害を伴う2型糖尿病患者における長期投与試験)
4体重に対する影響(参考情報)
ルセフィ2.5mgの腎機能障害(中等度)を伴う2型糖尿病患者への投与において、ベースラインからの体重変化量は二重盲検治療期Ⅰ終了時※1で-1.29kgであり、投与52週※2で-2.01kgであった。
■二重盲検治療期Ⅰ終了時および52週におけるベースラインからの体重変化量
二重盲検治療期Ⅰ終了時※1
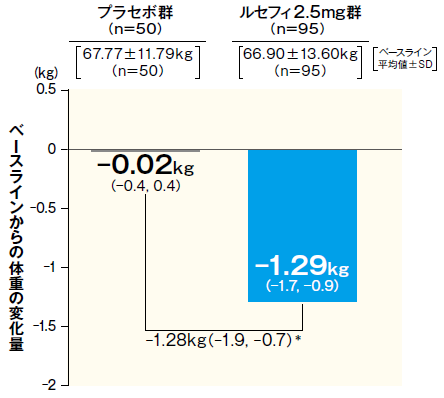
- FAS 最小二乗平均値(95% CI)
- *:p < 0. 001(vs. プラセボ群)、2標本t検定
投与52週
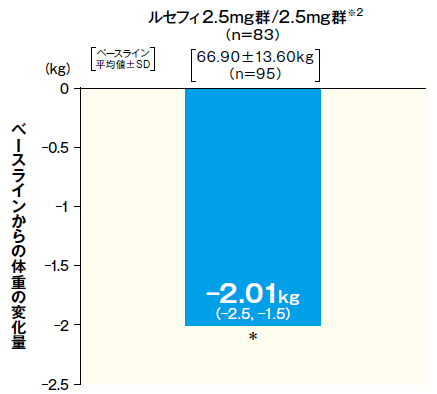
- FAS 平均値(95%CI)
- *:p < 0. 001(vs. ベースライン)、1標本t検定
- ※1 中止、脱落例などは、最終観察時点の測定値を解析。
- ※2 投与24週まで二重盲検下でルセフィ2.5mgを投与し、引き続き、非盲検下でルセフィ2.5mgを投与した症例。投与24週以降にルセフィ5mgへ増量した症例を含む。
- 8. 重要な基本的注意(抜粋)
- 8.3 本剤投与により、血清クレアチニンの上昇又はeGFR の低下がみられることがあるので、腎機能を定期的に検査するとともに、腎機能障害患者における治療にあたっては経過を十分に観察すること。[5.3 、5.4 、9.2 参照]
- 9.2 腎機能障害患者
- 9.2.1 重度の腎機能障害(eGFRが15mL/min/1.73m2以上29mL/min/1.73m2以下)のある患者又は透析中の末期腎不全患者
投与しないこと。本剤の効果が期待できないため。[5.3 、8.3 、16.6.1 参照] - 9.2.2 中等度の腎機能障害(eGFRが30mL/min/1.73m2以上59mL/min/1.73m2以下)のある患者
投与の必要性を慎重に判断すること。本剤の効果が十分に得られない可能性がある。[5.4 、8.3 、16.6.1 、17.1.3 参照]


